马强,张霞*,吴敬医,丁彩云,谷冰雨,徐四娟
(1.皖南医学院弋矶山医院超声医学科,安徽 芜湖 241001;
2.重症医学科;
3.妇产科)
甲状腺癌作为最常见的内分泌恶性肿瘤,患病率逐年增加,其中甲状腺乳头状癌(PTC)是最常见的亚型[1]。PTC的进展和整个发生发展过程涉及到基因改变,引起细胞增殖、细胞生长、血管生成和分化损失[2-3]。其中肿瘤微环境在PTC的侵袭和转移过程中发挥关键作用[1]。为探究PTC肿瘤微环境相关基因的作用机制,我们研究了癌症基因组图谱(TCGA)项目成果,进行了全面和多平台的方法来分析496个PTC的多维分子数据(不包括差和未分化的癌细胞,汇编所有肿瘤基因的改变),并将其与临床相关参数联系起来[4-5]。本文通过生物信息学方法对TCGA中PTC进行数据挖掘及分析,找出与PTC肿瘤微环境免疫相关的关键基因,为临床上从基因上预测甲状腺癌的临床发展状态,以及为治疗各期甲状腺癌提供新的靶点。
1.1 数据库PTC患者的基因表达谱及临床资料(如性别、年龄、分期、等级、生存率和最终结局)可从TCGA数据库(https://portal.gdc.cancer.gov/)获得。PTC患者的免疫评分和基质评分通过使用R软件(R Foundation for Statistical Computing,Vienna,Austria)中的ESTIMATE包来计算。根据免疫、基质评分的中位值,将所有患者分别分为高、低免疫细胞评分组以及高、低基质细胞评分组。
1.2 鉴定差异基因分别对基质及免疫细胞高、低分组,使用limma R软件包根据以下临界值识别出基质细胞和免疫细胞中差异表达基因:错误发现率(FDR)<0.05和|log2倍数变化(FC)|>2。再分别将基质细胞打分中上调、下调基因与免疫细胞打分中上调、下调基因取交集,得出肿瘤微环境相关的差异基因(DEGs)。
1.3 DEGs的GO和KEGG富集分析为了解DEGs在THCA中作用机制以及潜在生物学功能,在R软件中使用clusterProfiler软件包[6]进行GO分析[7]和KEGG富集分析[8],包括生物学过程(BP)、分子功能(MF)和细胞成分(CC),P<0.05为差异有统计学意义,最后将结果用ggplot2软件包绘制气泡图和圈图。
1.4 PPI网络的建立与模块分析为了了解不同基因之间的相互作用,使用STRING(https://stringdb.org/)在线数据库[9]分析DEGs的蛋白质-蛋白质相互作用(PPI)网络,并考虑了临界标准(最低要求互动得分>0.90)。使用3.8.0版(https://cytoscape.org)的cytoscape重建该网络,并使用分子复杂检测(MCODE)插件以识别PPI网络中的所有模块。
1.5 核心基因的鉴定与临床相关性分析使用cytoHubba(一个Cytoscape插件)中的度数算法[10]选择了排名前3位的核心基因,并与单因素Cox回归分析结果取交集得出差异的核心基因,并从中选择部分基因行单基因分析,通过R软件分析该基因在正常样本与甲状腺癌肿瘤样本间的表达差异,以及与甲状腺癌的预后以及临床分期的相关性。
1.6 GSEA富集分析借助GSEA 4.0.3软件对核心基因进行GSEA富集分析。
1.7 免疫细胞浸润的差异分析及相关性分析先对下载的基因表达数据进行归一化和过滤。然后利用CIBERSORT算法推断甲状腺癌和正常甲状腺中22种侵袭性免疫细胞的丰度,包括B细胞、树突状细胞、嗜酸性粒细胞、巨噬细胞、肥大细胞、单核细胞、中性粒细胞、自然杀伤(NK)细胞、浆细胞、T细胞及其不同亚型。通过使用蒙特卡罗采样,CIBERSORT得到了每个样本的P值,这为结果提供了一个置信度度量。以核心基因分组分别得出各免疫细胞与核心基因的相关性,绘制小提琴图以及散点图。
2.1 免疫评分和基质评分与临床参数显著相关我们从TCGA数据库下载了496例PTC患者的基因表达谱和临床信息。通过R中的ESTIMATE包对这些患者的免疫和基质评分进行匹配。共获得了510例具有完整免疫和基质评分的样本;
基质评分从-1 709.75至1 617.75,免疫评分从-1 298.62至3 244.00。使用中位免疫、基质评分作为临界值,将所有PTC样本分为高、低免疫评分组和高、低基质评分组。使用R中的生存软件包对患者免疫或基质评分与总体生存率之间的关系进行分析,结果显示,高、低免疫和基质评分组患者预后差异无统计学意义。此外,样本免疫和基质总评分与临床参数之间的关系分析发现,两者的总评分在不同的临床参数(临床分级、TNM分期)中存在显著的相关性(图1)。
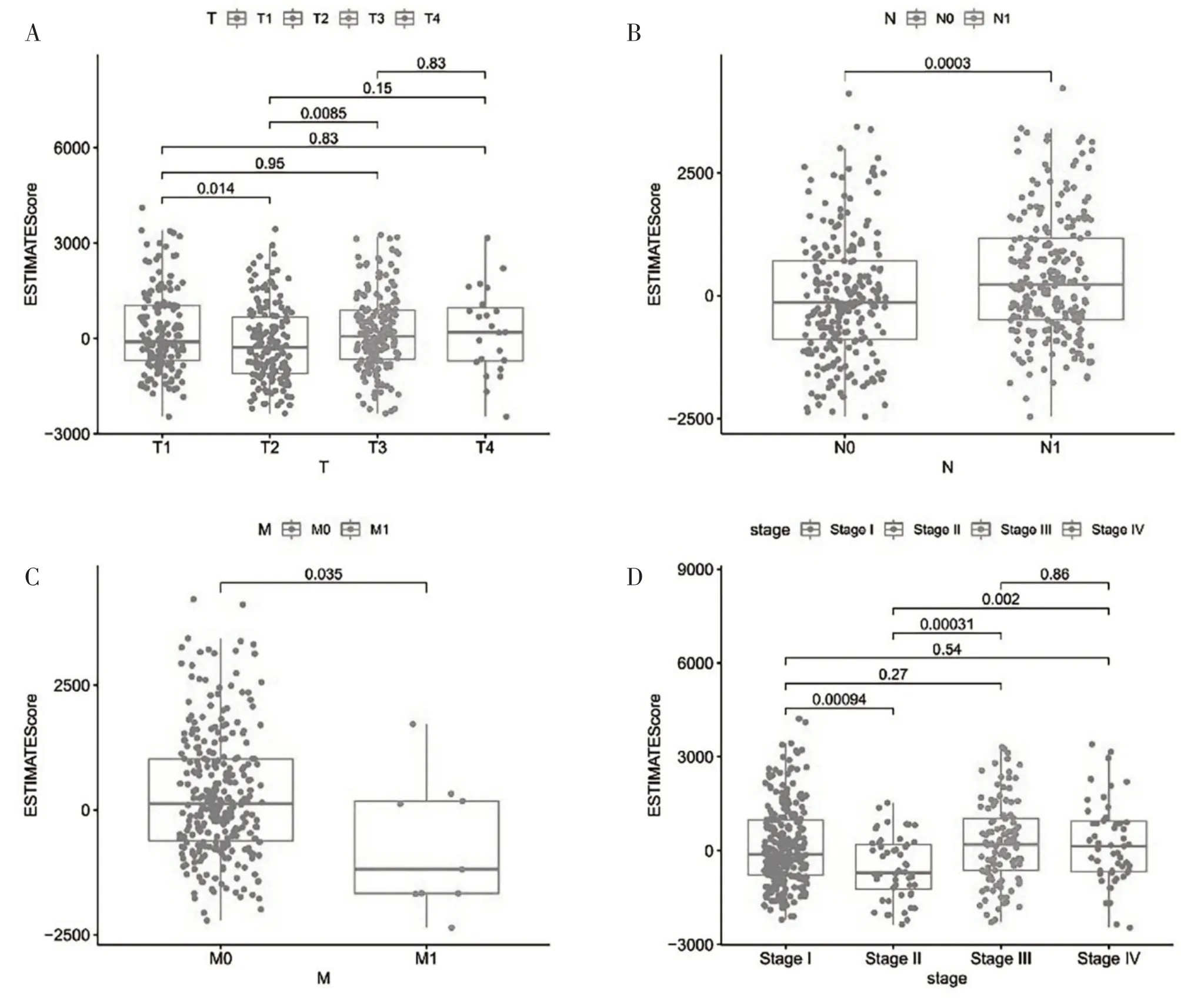
图1 免疫、基质总评分与临床参数的相关性分析散点图
2.2 鉴定DEGs为了更好地了解基因表达与免疫、基质评分之间的相关性,我们分析了510个PTC样本的基因表达谱。对于免疫评分,高分组与低分组相比,上调了690个基因,下调了60个基因(|log2倍数变化(FC)|>2,FDR<0.05)。对于基质评分,高分组与低分组相比,495个基因上调,8个基因下调(|log2倍数变化(FC)|>2,FDR<0.05)。分析免疫和基质评分的高分组中共同的上调和下调基因,发现了434个上调和7个下调的基因。获得的这441个基因被视为DEGs,并应用于进一步分析。
2.3 DEGs的GO功能和KEGG途径富集分析为了解这441个DEGs与PTC相关性的潜在机制,使用R软件对DEG进行了GO功能注释和KEGG途径富集分析。DEGs的GO功能分析分为生物过程(BP)、分子功能(MF)和细胞成分(CC),结果分析表明,主要的BP富集通路是体液免疫反应。在CC方面,免疫球蛋白复合物通路富集显著。对于MF,最显著的为抗原结合通路(图2A)。KEGG途径富集分析的结果表明,DEGs主要富集于包括细胞因子-细胞因子受体相互作用、造血细胞谱系、病毒蛋白与细胞因子及其受体的相互作用、趋化因子信号通路(图2B)。

图2 DEGs的GO功能和KEGG途径富集通路图
2.4 DEGs的PPI网络的构建和核心基因的鉴定为了进一步探讨DEGs及其调控机制,我们利用STRING在线数据库分析了DEGs,并构建了PPI网络。在PPI网络中,我们确定了139个基因(图3A)和7个功能模块(使用MCODE功能)。为更好地鉴定PTC微环境的核心基因,我们使用了Cytoscape软件中的cytohubba度数算法进一步分析了DEGs的PPI网络。根据它们的程度分数筛选了16个核心基因。排在前3位的核心基因如下:SAA1、CXCL10、CXCL11(图3B)。
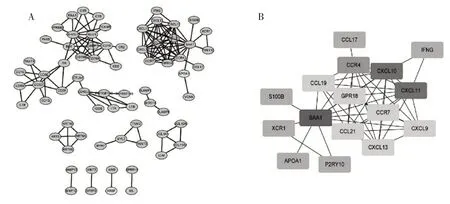
图3 DEGs的PPI网络构建(A)和核心基因的鉴定(B)
2.5SAA1单基因分析在下载的样本数据中对筛选出的3个核心基因分别进行表达差异分析,发现唯有SAA1存在表达差异,P=0.002 7(图4A)。对SAA1进行临床相关性的分析,包括性别、年龄、TNM分期以及分级等临床信息,发现SAA1在PTC不同的临床状态如肿瘤的大小、淋巴结转移以及肿瘤的分级存在较显著的差异,当SAA1高表达时,PTC患者更易发生淋巴结转移以及处于更高的分期。然而生存分析显示SAA1与PTC的预后无明显相关性(图4),此结果可能与PTC患者通常预后良好,生存率较高有关。
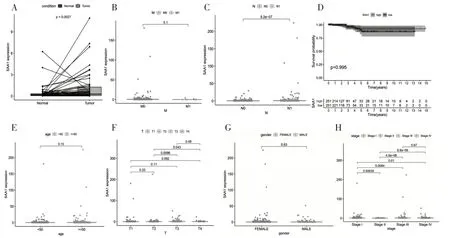
图4 SAA1临床相关性的箱线图与生存曲线分析
2.6 GSEA富集分析按SAA1表达水平的中位数将样本分为SAA1高表达组和SAA1低表达组,分别导入GSEA 4.0.3软件进行GSEA富集分析,结果表明,SAA1高表达样本中有22个富集通路(P<0.05),选择其中与免疫相关密切的抗原加工提呈通路、细胞黏附分子通路、趋化因子信号通路、细胞因子相互作用通路等9个通路绘制GSEA富集分析图(图5)。结果表明,免疫相关通路与SAA1高表达的肿瘤样本呈正相关。
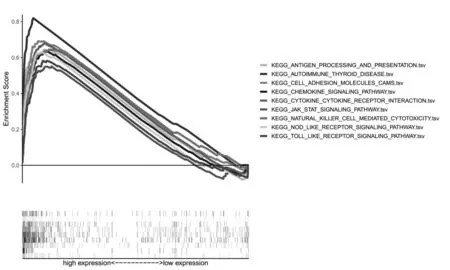
图5 GSEA富集分析图
2.7 免疫细胞浸润的相关性分析通过上述分析结果可得出肿瘤微环境的核心基因SAA1作用于免疫机制影响甲状腺癌的临床状态。
为了进一步研究通过哪些免疫细胞发挥效能,我们对TCGA数据进行归一化和过滤。然后利用CIBERSORT算法推断甲状腺癌和正常甲状腺中22种侵袭性免疫细胞的丰度(图6A)以及免疫细胞之间的相关性(图6B)。再根据SAA1的表达计算各个免疫细胞在SAA1高表达组和低表达组中的含量,结果发现:CD4记忆T细胞、调节性T细胞、γ-δ T细胞、静息的树突状细胞、活化的树突状细胞、静息的肥大细胞差异存在统计学意义(图6C)。
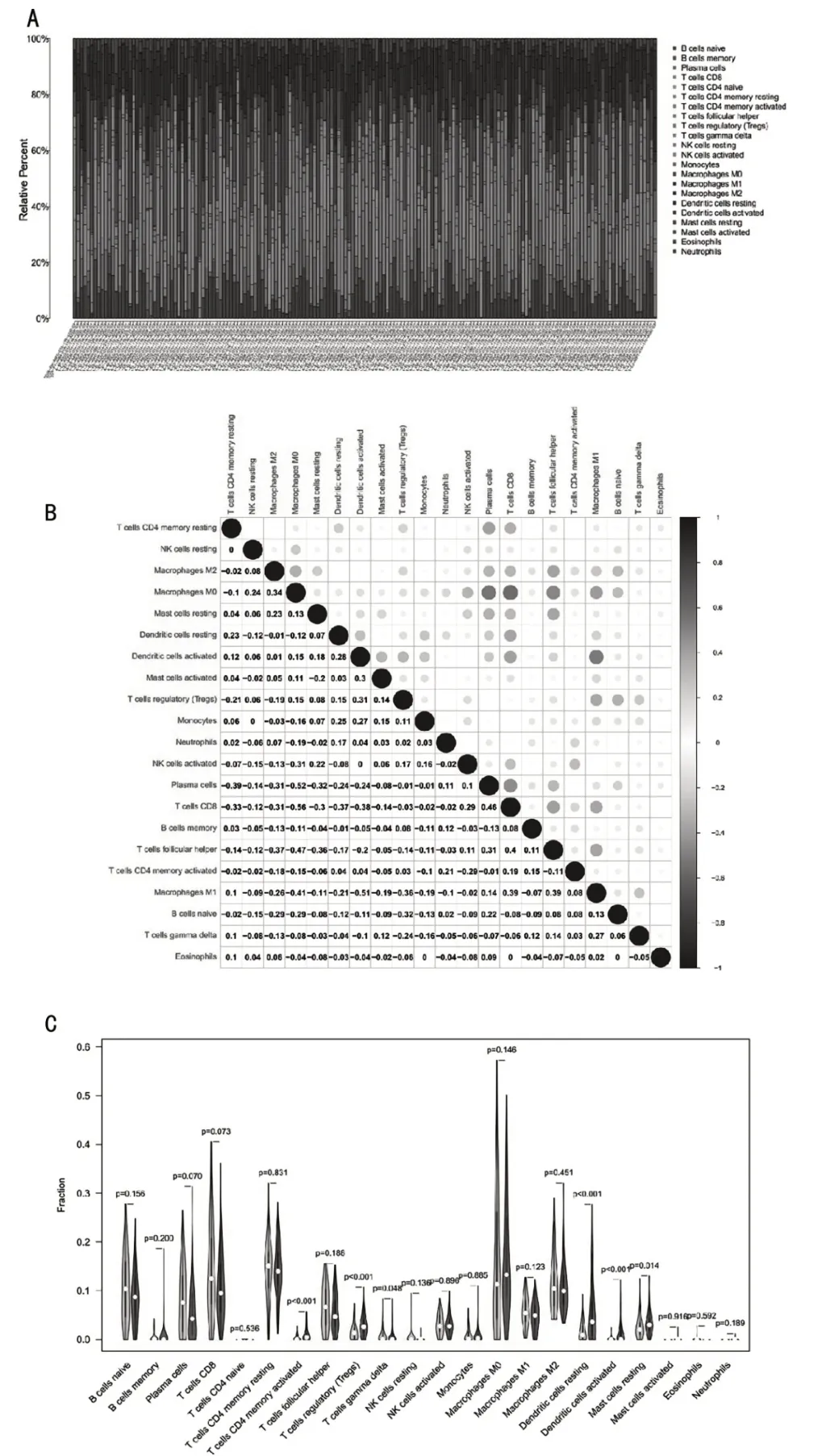
图6 免疫细胞相关性分析图以及SAA1与免疫细胞的小提琴图
2.8 作用免疫细胞将SAA1基因高、低表达分组后,免疫细胞含量存在差异;
通过spearman方法检验SAA1与各个免疫细胞的相关性,再将两者得出的免疫细胞取交集可得出,5个SAA1作用的免疫细胞。分别为CD4记忆T细胞、调节性T细胞、γ-δ T细胞、静息的树突状细胞、活化的树突状细胞。
甲状腺癌是最常见的内分泌腺癌,PTC是最常见的甲状腺癌。由于目前对各种形式的甲状腺癌遗传背景的了解还远远不够,因此大多数研究都集中在遗传基础上[11]。目前已有PTC生物信息学分析,结果显示在GEO平台中数据挖掘分析发现LPAR5、TFPI和ENTPD1与PTC患者的发展和预后相关[12],这些基因指标常与免疫浸润相关。炎症目前被认为是恶性肿瘤的重要组成部分[13]。如今肿瘤微环境已吸引了肿瘤研究界的关注,并在肿瘤发生和发展、治疗和预后中已被确认为一个关键因素[14-16]。越来越多的证据表明,肿瘤微环境的各种成分,如免疫细胞、可溶性因子和改变的细胞外基质,都对癌症的进展有积极作用[17]。而与肿瘤微环境相关的基因与甲状腺癌之间的联系尚未得到充分阐明。
本文采用生物信息学方法研究TCGA数据库中PTC基因表达谱及临床信息与肿瘤微环境之间的联系。首先使用ESTIMATE算法计算出样品中免疫、基质细胞的评分以及对临床状态如TNM分期等进行相关性分析,发现肿瘤微环境影响了PTC的发病,并作用于PTC的临床分期。本研究使用limma R软件包识别基质细胞高、低分组与免疫细胞高、低分组中DEGs,并对免疫、基质细胞高评分组和低评分组取交集得出441个DGEs。GO和KEGG分析得出主要的细胞因子受体相互作用等富集通路。进一步分析了DEGs的PPI网络,得出了SAA1等16个基因为重要连接站点。并对连接数前三的基因SAA1、CXCL10、CXCL11进行单基因分析发现,唯有SAA1在TCGA正常样品与肿瘤样品中的表达差异存在统计学意义,在配对的肿瘤样品和癌旁组织中,SAA1在肿瘤样品中表达量显著增高(P=0.002 7)。为进一步研究SAA1作用机制,我们对其行GSEA富集分析,发现结果里有22条通路上调,其中9条通路与免疫密切相关。进一步分析发现,SAA1可能通过影响免疫通路以及CD4记忆T细胞、调节性T细胞、γ-δ T细胞、静息的树突状细胞、活化的树突状细胞,最终影响甲状腺癌患者的临床状态。
目前研究表明SAA1在胰腺癌的发生发展中具有重要的调节作用[18]。高表达的SAA1具有作为晚期肾透明细胞癌患者的诊断和预后生物标志物的潜力,其可能作为晚期肾透明细胞癌患者的新型治疗靶点[19]。另外有研究表明SAA1可介导NOX4/ROS通路并激活p38MAPK/NF-κB通路,促进炎症介质的释放[20]。SAA1与机体的炎症反应存在一定关联,这与本研究的结果相符。本研究发现SAA1与PTCTNM分期及临床分级具有相关性,而与年龄、性别以及预后相关性无统计学意义。PTC肿瘤通常生长缓慢,且结合手术、甲状腺激素和放射性碘治疗效果较好,故PTC的预后一般都较好[21],且在TCGA数据库中的PTC数据是排除恶性度较高的癌症样本,所以可能会导致最终结果显示SAA1对于PTC的预后并不存在预测价值。但研究表明一旦PTC恶化,更具侵袭性和致命性,故若想预测PTC的预后,仍需对PTC发展进行更深入的机制研究,也可联合对比增强造影超声来预测甲状腺癌预后[22]。另外关于SAA1如何通过免疫反应通路影响PTC的发生发展仍需要更进一步的实验室研究。
本文的局限性在于只纳入了TCGA数据库中PTC数据,没有与其他数据库进行联合比较。本研究结果表明肿瘤免疫微环镜对于PTC的发生发展具有重要意义,并发现了SAA1作为PTC潜在的免疫浸润的生物标志物,在未来将有助于PTC的诊断、预后和靶向治疗的进一步临床应用。
猜你喜欢 甲状腺癌基质通路 甲状腺癌“低位领”式与“L”型切口淋巴结清扫术的比较承德医学院学报(2022年2期)2022-05-234种不同配比基质对戈壁日光温室西瓜生长及产量的影响农业科技与信息(2021年21期)2021-12-28机插秧育苗专用肥——机插水稻育苗基质中国土壤与肥料(2021年5期)2021-12-12金银花扦插育苗基质复配及验证中国土壤与肥料(2021年5期)2021-12-02小檗碱治疗非酒精性脂肪肝病相关通路的研究进展现代临床医学(2021年4期)2021-07-31Wnt/β-catenin信号转导通路在瘢痕疙瘩形成中的作用机制研究云南医药(2021年3期)2021-07-21分化型甲状腺癌切除术后多发骨转移一例国际放射医学核医学杂志(2021年10期)2021-02-28分化型甲状腺癌肺转移的研究进展国际放射医学核医学杂志(2021年10期)2021-02-28白芍总苷调控Sirt1/Foxo1通路对慢性心力衰竭大鼠的保护作用研究天津医科大学学报(2021年1期)2021-01-26护理干预在降低甲状腺癌患者焦虑中的应用研究中华养生保健(2020年4期)2020-11-16